A multidisciplinary team of researchers at Waterloo’s Cheriton School of Computer Science and Western University has developed a computational method that within minutes can identify and classify viruses such as SARS-CoV-2, the respiratory pathogen responsible for the COVID-19 pandemic.
The team used an alignment-free method coupled with iterative rounds of machine learning to identify SARS-CoV-2 and to determine its taxonomic relationship to other viruses. Unlike alignment-based approaches, the novel method requires no specialized biological knowledge of the organism being assessed or specific genetic annotation, and it can be used to both identify and classify any unknown species with a high degree of accuracy.
“Identifying the COVID-19 virus and determining its relationship to other coronaviruses using alignment-based methods has been described in several recent papers,” said Lila Kari, University Research Professor at the Cheriton School of Computer Science and an expert in biomolecular computation. “The difference in our research is that we used an alignment-free method, coupled with machine learning, to identify the COVID-19 virus and classify its relationship to other viruses within mere minutes.”
Lila Kari is a Professor and University Research Chair in the Cheriton School of Computer Science. Author of more than 200 peer-reviewed articles, she is regarded as one of the world’s experts in the area of biomolecular computation — using biological, chemical and other natural systems to perform computations.
Alignment-based classification approaches examine several gene sequences and then they compare the degree of similarity across several species, Professor Kari said. “These methods are successful in finding sequence similarities and hence genetic relationships, but they have drawbacks. The first is that the gene you’re examining has to exist in every organism in which you are comparing it. The second is that alignment requires genetic sequences to be contiguous and homologous — in other words, inherited from a common ancestor — which greatly restricts the type of genetic fragments that can be used for analysis. The third is that the comparison takes a lot of time. Let’s assume a particular virus gene is 3,000 base pairs or ‘genetic letters’ long. If you want to compare that sequence of base pairs across the 5,500 or so known species of viruses it would be extremely difficult because of the heavy computation time required.”
In contrast, the alignment-free method she and her team developed allows researchers to identify and classify novel pathogens accurately and quickly. “Within minutes, a few hours at most, we can determine what species a given pathogen is and to which other pathogens it is most closely related — critical taxonomic information that when coupled with already known concerns could alert us as to how alarmed we should be.”
Early identification and classification of viruses and other pathogens are increasingly important in a globalized world. SARS-CoV-2, the virus that causes COVID-19, is the third highly pathogenic coronavirus affecting humans this century alone, after the Middle East respiratory syndrome coronavirus (MERS-CoV), which caused a disease outbreak beginning in 2012, and the Severe Acute Respiratory Syndrome coronavirus (SARS-CoV or SARS-CoV-1), which caused a disease outbreak beginning in 2002.
Knowing what species a virus is and to which other viruses it is related not only speeds development of treatments and vaccines, but it also informs the urgency of public health measures to help contain its spread. Since the event that introduced SARS-CoV-2 to humans in late 2019, as of April 27, 2020 the world has seen more than 3 million confirmed cases of COVID-19 and more than 210,000 deaths attributed to it, according to the COVID-19 Dashboard developed by the Center for Systems Science and Engineering at Johns Hopkins University.
With expertise spanning computer science, genetics and statistics, Professor Kari and her team began by downloading the genetic sequences of the more than 5,500 virus species catalogued in the National Center for Biotechnology Information database. Instead of using the genetic sequences of the viruses directly as alignment-based methods do, the researchers extracted a unique characteristic from each virus’s genome and compressed it mathematically into what they call a genomic signature — a unique numerical representation of the virus’s genetic sequence.
“This approach has three significant advantages, the first being speed,” Professor Kari explained. “Instead of comparing a number of gene sequences across organisms letter by letter, our alignment-free approach can quickly compare the genomic signature of one organism to that of other organisms. The second is that alignment-free approaches have a high degree of generality — you can literally compare any organism to any other organism. The third advantage is that the technique is comprehensive. No information is lost when the genetic sequences are distilled mathematically. The resulting genomic signature retains a general but significant characteristic of the species from which it is derived, one that can be used to compare with the genomic signatures of other species at large scale.”
Using the genomic signatures, the researchers employed machine learning and something akin to the 20 questions game — a series of questions that become ever more specific that allow a clever questioner to get ever closer to an answer. The twist here is that the 20 questions game is what mathematicians call a decision tree, a kind of decision-support tool that employs a branching-like model of decisions and possible consequences, combined with power of machine learning to drill down from the highest level of taxonomic classification to the very specific.
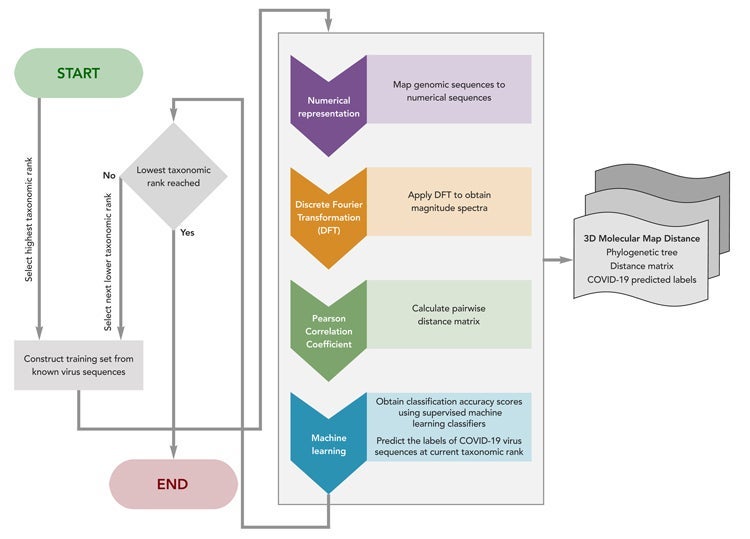
Machine
learning
combines
with
digital
signal
processing,
resulting
in
successions
of
increasingly
accurate
classifications
of
the
bare-bones
COVID-19
viral
genome.
Download
a
larger
version
of
this
illustration —
jpeg
and
PDF.
{kind=link}
Using this iterative method, shown conceptually in the figure above, the researchers classified SARS-CoV-2 as a virus belonging to the family Coronaviridae (the family of all coronaviruses) and more specifically to the genus Betacoronavirus (one particular genus of coronavirus that infects mammals). These results were obtained with 100 percent accuracy as well as provided confirmation that SARS-CoV-2 is most closely related to three other coronaviruses found in bats.
“One wonders, had we known during the very early days of the COVID-19 outbreak that the pathogen causing pneumonia and other respiratory symptoms in Wuhan, China was closely related to the coronavirus that caused the deadly SARS outbreak in the early 2000s and that our results support a bat origin, it might have set off alarm bells quickly and loudly across the globe,” Professor Kari said. “And, importantly, we would have had a convincing argument early on to implement public health measures to help contain its spread.”
The paper detailing this study, titled “Machine learning using intrinsic genomic signatures for rapid classification of novel pathogens: COVID-19 case study,” was written by University Research Professor Lila Kari and her colleagues — Gurjit S. Randhawa (a PhD student in Western’s Department of Computer Science), Maximillian P.M. Soltysiak and Hadi El Roz (both undergraduate students in Western’s Department of Biology), Camila P.E. de Souza (Assistant Professor in Western’s Department of Statistical and Actuarial Sciences) and Kathleen A. Hill (Associate Professor in Western’s Department of Biology). Their open-access peer-reviewed paper was published on April 24, 2020 in the journal PLOS ONE.
Citation: Randhawa GS, Soltysiak MPM, El Roz H, de Souza CPE, Hill KA, Kari L (2020). Machine learning using intrinsic genomic signatures for rapid classification of novel pathogens: COVID-19 case study. PLOS ONE 15(4): e0232391. https://doi.org/10.1371/journal.pone.0232391.